At WiseGEEK, we're committed to delivering accurate, trustworthy information. Our expert-authored content is rigorously fact-checked and sourced from credible authorities. Discover how we uphold the highest standards in providing you with reliable knowledge.
What is Thalassemia?
Thalassemia is a serious disorder that affects the physical presentation and composition of hemoglobin in an individual’s blood. Recognized as a genetic condition, thalassemia carries a high mortality rate often necessitating regular blood transfusions to increase one’s life expectancy. Aside from blood transfusions, treatment for this condition may also require the application of chelation therapy and bone marrow transplant. If left untreated, this condition may increase one’s susceptibility to infection, organ failure, and anemia.
Many who acquire this disorder may exhibit a yellowed skin tone, a condition known as jaundice, due to the presence of elevated levels of bilirubin in their system caused from the thalassemic-induced assault on their red blood cells. Often experiencing signs of illness soon after birth, infants with this disorder may demonstrate facial deformity and present impaired growth. Additional signs of this blood disorder include chronic fatigue, persistent headaches, and shortness of breath.

In the presence of this inherited autosomal recessive blood disease, oxygen-carrying hemoglobin demonstrates an abnormal physical presentation as well as chemical composition. Under normal circumstances, the protein hemoglobin is composed of both beta and alpha globin; however, in the presence of thalassemia one of the globins is either missing or mutated. The chromosomal gene mutation damages and eliminates red blood cells causing the onset of anemia.

The condition is generally diagnosed as either major or minor in presentation, depending on the severity of symptom presentation and whether the individual inherited the defective gene from one or both parents. As with many inherited conditions, such as sickle-cell anemia, an individual may inherit the mutated gene from one parent and remain a carrier or inherit a defective gene from each parent and become symptomatic. Those who remain a carrier often remain asymptomatic, meaning they experience no symptoms at all.

There are several diagnostic blood tests that may be performed to confirm the presence of the disorder. Generally, a physical examination will detect abdominal distention resulting from spleen inflammation, often prompting the administration of a battery of blood tests to evaluate the presentation of hemoglobin in the individual’s blood. Tests performed frequently include a complete blood count (CBC) and hemoglobin electrophoresis.

Thalassemia is known to cause premature death in young adults. Individuals with this condition are often more susceptible to serious complications, including infection and organ failure. Frequently manifesting during young adulthood, such complications may be deterred with proper treatment that is generally centered on the regular administration of blood transfusions. Some individuals may also undergo chelation therapy and bone marrow transplant to help stabilize their condition and manage symptoms. Complications associated with thalassemia often include increased susceptibility to infection and organ damage due to the prolonged presence of elevated iron levels.
AS FEATURED ON:
AS FEATURED ON:

-
![Symptoms of thalassemia may include chronic fatigue and headaches.]() By: NOBUSymptoms of thalassemia may include chronic fatigue and headaches.
By: NOBUSymptoms of thalassemia may include chronic fatigue and headaches. -
![Blood tests may be conducted to diagnose thalassemia.]() By: kastoBlood tests may be conducted to diagnose thalassemia.
By: kastoBlood tests may be conducted to diagnose thalassemia. -
![Thalassemia may cause premature death in young adults.]() By: ryflipThalassemia may cause premature death in young adults.
By: ryflipThalassemia may cause premature death in young adults. -
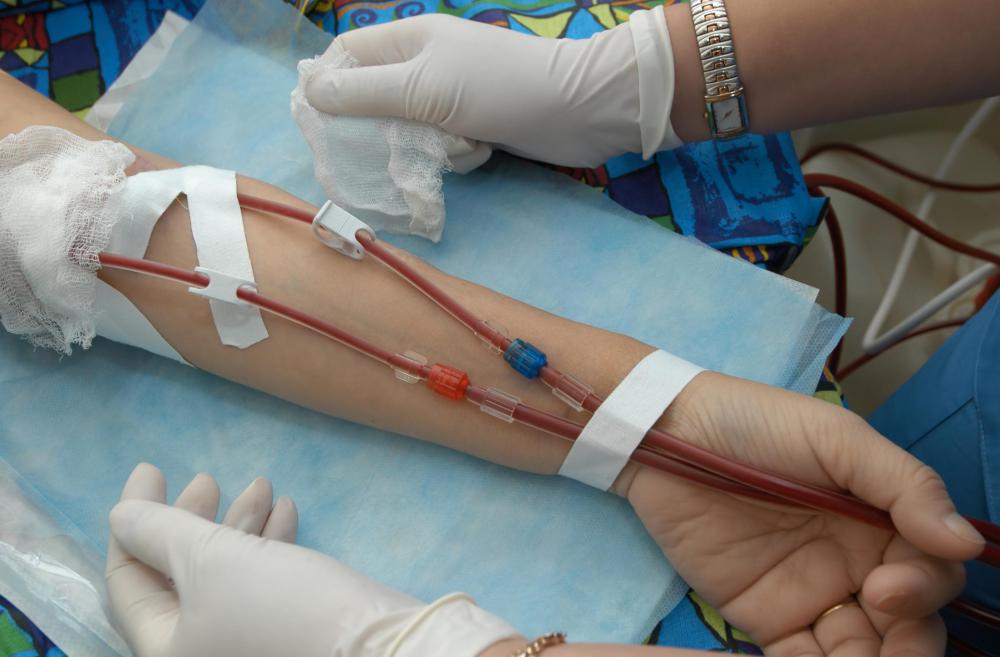
![Thalassemia may be deterred with proper treatment that is generally centered on the regular administration of blood transfusions.]() By: Max TacticThalassemia may be deterred with proper treatment that is generally centered on the regular administration of blood transfusions.
By: Max TacticThalassemia may be deterred with proper treatment that is generally centered on the regular administration of blood transfusions. -
![Shortness of breath is one symptom of thalassemia.]() By: nuiikoShortness of breath is one symptom of thalassemia.
By: nuiikoShortness of breath is one symptom of thalassemia.




Discuss this Article
Post your comments